Introducció
La cinemàtica és una branca de la física mecànica que estudia el moviment dels objectes i l’anàlisi de les trajectòries que formen. Si la dimensió de l’objecte és molt més petita que la de la trajectòria, el tractem com un punt o una partícula sense dimensions. Per exemple, considerarem una molècula com una partícula en moviment, però no seria adequat tractar les marees com a partícules si en volguéssim estudiar el moviment.
El moviment és un canvi de posició en el temps. Pot ser rectilini o circular (de translació, de rotació o de vibració) en una, dues o tres dimensions i és relatiu a l’observador. L’expressarem en coordenades cartesianes, esfèriques o cilíndriques (tot i que en mecànica relativista s’usa la geometria hiperbòlica) per tal que els càlculs siguin més senzills.
La trajectòria d’una partícula és el camí que segueix mentre es mou i està formada per moviments lineals i curvilinis.
En aquesta entrada estudiarem el moviment lineal i circular en una i dues dimensions fent ús del càlcul vectorial i escalar. Les magnituds del moviment lineal són: la posició, la velocitat, l’acceleració i el temps. Les tres primeres són magnituds vectorials, però la darrera és escalar.
En les equacions escalars, el signe de les magnituds vectorial indica el sentit del vector segons el criteri de signes dels eixos de coordenades (amunt, dreta: positiu, avall, esquerra: negatiu). Dos vectors de sentits diferents es resten, mentre que si tenen el mateix, se sumen. En conseqüència, si el vector velocitat i el vector acceleració tenen el mateix sentit, la partícula accelerarà, però si tenen sentit diferent, frenarà.
Per a resoldre un exercici de cinemàtica, cal escriure abans de res les equacions de cada partícula. A continuació, farem els càlculs que calguin per a respondre les qüestions de l’exercici.
2.1 Posició
La posició d’una partícula són les coordenades del punt (x,y) on és en un instant mesurades des de l’origen del sistema de referència triat.
Quan una partícula canvia de posició \((\vec{x})\) diem que ha transcorregut un temps (»t»).
\(
\displaystyle{
\vec{r}=x \vec{i}+y \vec{j}+z \vec{k}\\
\Delta \vec {r}=\vec {{r}_{2}}-\vec {{r}_{1}}\\
}
\)
El canvi de posició d’una partícula és el »desplaçament» \((\Delta \vec{r}=\vec{x}_{2}-\vec{x}_{1})\) El mòdul d’aquest vector és la distància que ha recorregut (»d»).
Exemple:
\(
\displaystyle{
\vec{r_{1}}=\left(3\vec{i}-4\vec{j}+5\vec{k}\right)
\\
\vec{{r}_{2}}=\left(2\vec{i}+2\vec{j}-8\vec{k}\right)
\\
\mathrm{\Delta }\vec{r}=\vec{{r}_{2}}-\vec{{r}_{1}}=\left(2\vec{i}+2\vec{j}-8\vec{k}\right)-\left(3\vec{i}-4\vec{j}+5\vec{k}\right)=\left(-1\vec{i}+6\vec{j}-13\vec{k}\right)
\\
d=| \vec r|=\sqrt{(-1)^2+(+6)^2+(-13)^2}=\sqrt{206}
}
\)
2.2 Velocitat
La velocitat d’una partícula és el vector desplaçament dividit pel temps que triga a moure’s entre dos punts i ens indica la rapidesa amb la qual canvia de posició amb el temps. Per tant, és un vector que té la mateixa direcció i sentit que el vector desplaçament i sempre és tangent a la trajectòria en cada instant.
La velocitat mitjana és el desplaçament total que ha fet la partícula durant un període de temps, però no ens informa dels detalls del moviment. Per exemple, podria haver-se desplaçat a la mateixa velocitat durant tot el trajecte, o bé haver-se parat, frenat i accelerat.
\(\displaystyle{\vec{{v}_{m}}=\frac{\mathrm{\Delta }\vec{r}}{\mathrm{\Delta }t}}\)
Si aquesta partícula es mou amb una velocitat variable, ens caldrà determinar a quina velocitat es desplaça en cada instant de la trajectòria. Definim la velocitat instantània com el límit del vector desplaçament \(\mathrm{\Delta }\vec{r}\) en un interval de temps infinitament petit:
\(\displaystyle{\vec{{v}_{i}}=\underset{\mathrm{\Delta }\rightarrow 0}{\lim }\frac{\mathrm{\Delta }\vec{r}}{\mathrm{\Delta }t}=\underset{\mathrm{\Delta }\rightarrow 0}{\lim }\frac{\vec{r}\left(t+\mathrm{\Delta }t\right)-\vec{r}\left(t\right)}{\mathrm{\Delta }t}=\frac{\mathit{d \vec r}}{\mathit{dt}}}\)
El vector velocitat instantània és el pendent de la funció posició en un punt (vegeu gràfic de l’apartat 2.1).
Com que la velocitat és una magnitud vectorial, el canvi pot ser del mòdul, de la direcció o del sentit, però la direcció i sentit de \(\vec v_i\) serà la de \( \Delta \vec r\) quan els dos punts de posició estiguin infinitament a prop.
Per tant, com que \(\vec{r}=x\vec{i}+y\vec{j}+z\vec{k}\)
\(
\displaystyle{
\vec{{v}_{i}}=\frac{d\vec{r}}{\mathit{dt}}=\frac{\mathit{dx}}{\mathit{dt}}\vec{i}+\frac{\mathit{dy}}{\mathit{dt}}\vec{j}+\frac{\mathit{dz}}{\mathit{dt}}\vec{k}={v}_{x}\vec{i}+{v}_{y}\vec{j}+{v}_{z}\vec{k}
}
\)
2.3 Acceleració
L’acceleració és la rapidesa amb la qual canvia la velocitat (mòdul, direcció o sentit) d’una partícula amb el temps.
L’acceleració mitjana és la variació de velocitat de la partícula en un interval de temps. Consegüentment, té la mateixa direcció i sentit que la velocitat, però tampoc ens informa dels detalls del moviment durant el desplaçament.
\(\displaystyle{{a}_{m}=\frac{\mathrm{\Delta }\vec{v}}{\mathrm{\Delta }t}}\)
Però, quan l’acceleració és variable, per tal de determinar-la en un instant determinat del temps, definim l’acceleració instantània com el canvi de la velocitat en un instant infinitament petit de temps:
\(\displaystyle{\vec{a}=\underset{\mathrm{\Delta }t\rightarrow 0}{\lim }\frac{\mathrm{\Delta }\vec{v}}{\mathrm{\Delta }t}=\frac{d\vec{v}}{\mathit{dt}}}\)
i, per tant,
\(\displaystyle{
\vec a=\frac{dv_x}{dt}\vec i+\frac{dv_y}{dt}\vec j+\frac{dv_z}{dt}\vec k=a_x \vec i+a_y \vec j+a_z \vec k
}
\)
Exemple:
\(
\displaystyle{
\text{L’equació vectorial de desplaçament d’una partícula és:}
\\[0.5cm]
\vec{r(t)}=(5+12{t}^{2}) \vec{i}-2t \vec{j}+({t}^{3}+8t) \vec{k}
\\[0.5cm]
\text{a) Quina és la posició en els instants t=2 i 5 segons i quin ha estat el desplaçament?}
\\[0.5cm]
\vec{r(2)}=(5+12{(2)}^{2})\vec{i}-2(2)\vec{j}+(t{(2)}^{3}+8(2))\vec{k}=
\\
53\vec{i}-4\vec{j}+24\vec{k}
\\
\vec{r(5)}=(5+12{(5)}^{2})\vec{i}-2(5)\vec{j}+({(5)}^{3}+8(5))\vec{k}=305\vec{i}-10\vec{j}+165\vec{k}\\
\mathrm{\Delta }\vec{r}=\vec{r}(5)-\vec{r}(2)=(305\vec{i}-10\vec{j}+165\vec{k})-(53\vec{i}-4\vec{j}+24\vec{k})=252\vec{i}-6\vec{j}+141\vec{k}
\\[0.5cm]
\text{b) Quina és la velocitat en els instants t=3 i 5 i quina és la velocitat mitjana?}
\\[0.5cm]
\vec{v(t)}=d\frac{\vec{r(t)}}{\mathit{dt}}=\frac{d}{\mathit{dt}}[(5+12{t}^{2})\vec{i}-2t\vec{j}+({t}^{3}+8t)\vec{k}]=
\\
24t\vec{i}-2\vec{j}+(3{t}^{2}+8)\vec{k}\\
\vec{v(3)}=24(3)\vec{i}-2\vec{j}+\lbrack 3{(3)}^{2}+8\rbrack \vec{k}=
\\
72\vec{i}-2\vec{j}+35\vec{k}\\
\vec{v(5)}=24(5)\vec{i}-2\vec{j}+\lbrack 3{(5)}^{2}+8\rbrack \vec{k}=
\\
120\vec{i}-2\vec{j}+83\vec{k}\\
\mathrm{\Delta }\vec{v}=(120\vec{i}-2\vec{j}+83\vec{k})-(72\vec{i}-2\vec{j}+35\vec{k})=
\\
48\vec{i}+48\vec{k}\\
\vec{{v}_{m}}=\frac{\mathrm{\Delta }v}{\mathrm{\Delta }t}=\frac{48\vec{i}+98\vec{k}}{5-3}=\frac{48}{5}\vec{i}+\frac{48}{3}\vec{k}
\\[1cm]
\text{c) Calculeu el vector acceleració:}
\\[0.5cm]
a(t)=\frac{{d}^{2}\lbrack \vec{r}(t)\rbrack }{\mathit{dt}^{2}}=d\frac{\vec{v}(t)}{\mathit{dt}}=24\vec{i}+6t\vec{k}
}
\)
2. Moviment lineal en una dimensió
2.1 Horitzontal
Si »a=0», la velocitat és constant i el moviment és anomenat MRU o moviment rectilini uniforme.
Si »a ≠ 0» i és constant, el moviment és anomenat MRUA o moviment rectilini uniformement accelerat.
Si l’acceleració és constant, la velocitat mitjana \(\displaystyle{\bar{v}=\frac{v_f+v_0}{2}}\) en qualsevol interval també és constant.
\(
\displaystyle{
\text{Si substituïm } t=\frac{v_f-v_0}{a} \text{ a } x=x_o+\bar v t
\\
v_f^{2}-v_0^2=2a(x-x_0)
\\[1cm]
\text{I com que,}
\\[0.5cm]
\bar v= \frac {1}{2}(v_f+v_0) \text{ i }v_f=v_0+at
\\
x=x_0+\bar v t \rightarrow x=x_0+\frac {1}{2}(v_f+v_0)t
\\
x=x_0+\frac{1}{2}[(v_0+at)+v_0]t
\\
x=x_0+v_0t+\frac {1}{2}at^2
}
\)
Per tant, les equacions de moviment horitzontal són:
\(\displaystyle{\begin{array}{c}x={x}_{0}\pm {v}_{0}\cdot t\pm \frac{1}{2}\mathit{at}\mathrm{{^2}}\\
{v}_{f}={v}_{0}\pm \mathit{at}\end{array}}\)
Exemple:
En quina posició i temps es trobaran els mòbils següents?
\(
\displaystyle{
\text{1. Tranformem totes les dades SI:}
\\[0.5cm]
90\frac{{Km}}{h}\cdot \frac{1000\text{m}}{1\text{Km}}\cdot \frac{1\text{h}}{3600\text{s}}=25\text{m/s}
\\[0.5cm]
\text{1. Escrivim les equacions de moviment de cada partícula:}
\\[0.5cm]
{x}_{A}={x}_{0A}+{v}_{0A}t \rightarrow {x}_{A}=0+25t
\\
{v}_{{fA}}={v}_{0A}=25\text{m/s}
\\[0.5cm]
\text{Però, com que la partícula A haurà trigat en recórrer 100}m{\colon }
\\[0.5cm]
100=0+25t \rightarrow t=4\text{s}
\\[0.5cm]
\text{llavors,}
\\[0.5cm]
{x}_{A}=100+25(t-4)
\\
{x}_{B}={x}_{0B}+{v}_{0B}t+\frac{1}{2}{{at}}^{2}=0+0t+\frac{1}{2}3t{{^2}}=\frac{3}{2}{t}^{2}
\\
{v}_{{fB}}={v}_{0B}+{at}=0+3t=3t
\\[0.5cm]
\text{2. Com que la posició i el temps de trobada és el mateix per a ambdues:}
\\[0.5cm]
{x}_{A}={x}_{B} \rightarrow 100+25(t-4)=\frac{3}{2}{t}^{2} \rightarrow \frac{3}{2}{t}^{2}-25t=0
\\
t=16.\hat{6} \text{s} \rightarrow {x}_{A}={x}_{B}=\frac{3}{2}{(16.\hat{6})}^{2}={416.\hat 6}\text{m}
\\[0.5cm]
\text{I la velocitat de la moto serà: }{v}_{B}=3 \cdot (16.\hat{6})=3(16.\hat{6})={49.8}\text{m/s}
}
\)
La representació gràfica de la posició, la velocitat i l’acceleració en funció del temps de cada moviment és:
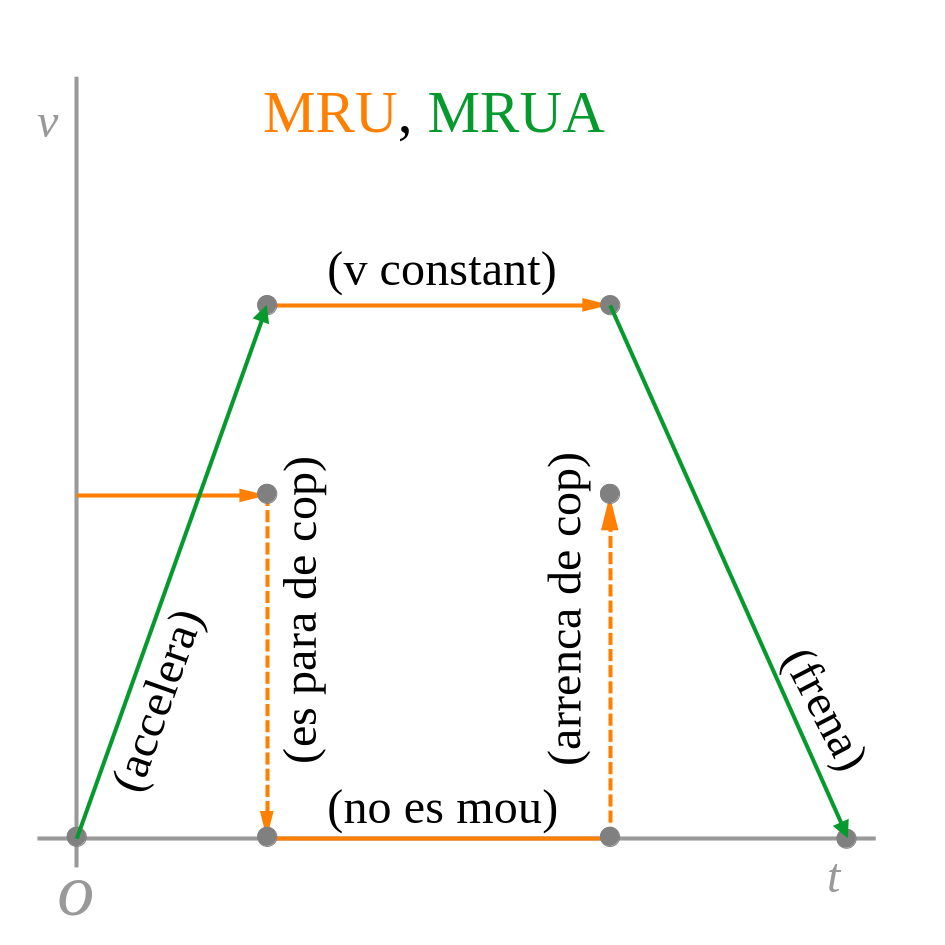
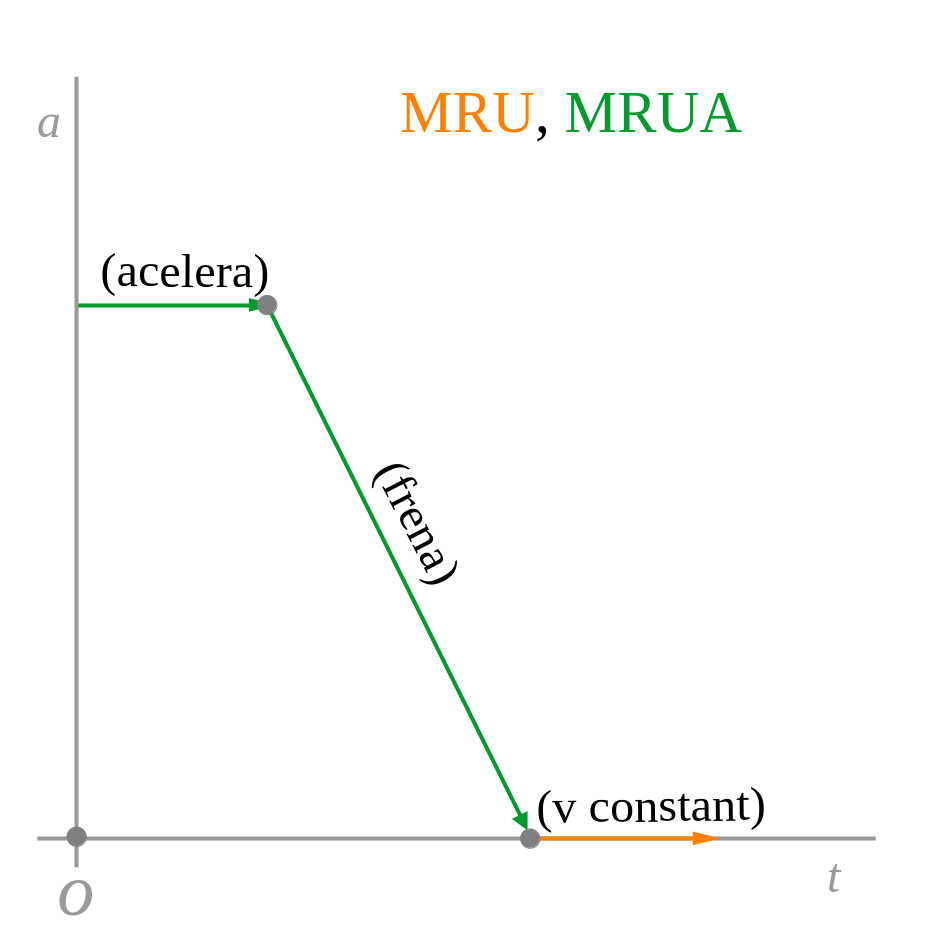
2.2 Vertical (caiguda lliure)
Les equacions de moviment de la caiguda lliure d’una partícula, són:
\(\displaystyle{
y={y}_{0}\pm {v}_{0}\cdot t-\frac{1}{2}g{t}^{2}
\\
{v}_{f}={v}_{0}-gt
}
\)
I, similarment al cas de moviment horitzontal:
\(\displaystyle{
{v}_f^{2}={v}_{0}^{2}+2a\cdot (y-y_0)}\)
Durant el camí de pujada i de baixada, canvia el mòdul i el sentit, però no pas la direcció del vector velocitat. La direcció, sentit i el mòdul del vector acceleració \(\vec{g}\) és sempre el mateix.
Exemples:
\(\displaystyle{
\text{1.Escrivim les equacions de moviment:}
\\[0.5cm]
y={y}_{0}+{v}_{0}t-\frac{1}{2}g{t}^{2}
\\
{v}_{f}={v}_{0}-gt
\\[0.5cm]
\text{2. Com que quan } t=2, {v}_{f}=+30\text{m/s}
\\[0.5cm]
{v}_{0}={v}_{f}+gt=30+9.8\cdot 2=49.6\text{m/s}
\\[0.5cm]
\text{i, quan arriba a dalt de tot:}
\\[0.5cm]
{v}_{f}=0={v}_{0}-gt=49.6-9.8t \rightarrow t=\frac{49.6.4}{9.8}=5.06\text{s}
\\[0.5cm]
\text{Per tant, l’alçada màxima que assoleix és:}
\\[0.5cm]
y={y}_{0}+{v}_{0}t-\frac{1}{2}9.8{t}^{2}=0+49.6 \cdot 5.06-4.9 \cdot {5.06}^{2}=125.52\text{m}
}
\)
Moviment en el pla (dues dimensions)
3.1 Moviment parabòlic
És un moviment en el qual el component horitzontal és un MRU (2.11) i el vertical un MRUA (2.1.2).
Hi ha tres punts de referència (A, B, C) que ens serviran per a plantejar les equacions de moviment i resoldre l’exercici:
\(\displaystyle{
A:
\\[0.5cm]
y=0,\\
{v}_{x}=v_0\cdot \cos \theta \enspace (\mathit{constant},\mathit{MRU}),\\
{v}_{y\mathit{m\grave{a}x}}={v}_{0}\cdot \sin \theta \enspace (\mathit{MRUA})
\\[0.5cm]
B\mathrm{\colon }
\\[0.5cm]
y_{\mathit{m\grave{a}x}}=h,\\
{v}_{x}={v}_{0}\cdot \cos \theta \enspace (\mathit{constant},\mathit{MRU}),\\
{v}_{y}=0(\mathit{MRUA})
\\[0.5cm]
C\mathrm{\colon }
\\[0.5cm]
y=0,\\
{v}_{x}=v\cdot \cos \theta (\mathit{constant},\mathit{MRU}),\\
{v_{y\mathit{m\grave{a}x}}}=-{v}_{0}\cdot \sin \theta \enspace (\mathit{MRUA})\\
R(\mathit{abast})={v}_{x}\cdot t
}
\)
Exemple:
\(\displaystyle{
\text{1. Les equacions de moviment, són:}
\\[0.5cm]
\text{a) Per al moviment vertical:}
\\[0.5cm]
y={y}_{0}\pm {v}_{0y}t-\frac{1}{2}g{t}^{2}
\\
{v}_{\mathit{fy}}={v}_{0y}-gt
\\[0.5cm]
\text{b. Per al moviment horitzontal:}
\\[0.5cm]
x={x}_{0}\pm {v}_{0x}t
\\[0.5cm]
{v}_{f}={v}_{0}
\\[0.5cm]
\text{2. Subtituint amb les dades que tenim:}
\\[0.5cm]
(i) \enspace y=40+30\cdot \sin (35)t-\frac{1}{2}9.8{t}^{2}
\\
(\mathit{ii}) \enspace {v}_{fy}=30\cdot \sin (35)-9.8t
\\
(\mathit{iii}) \enspace 20=0+30\cdot \cos (35)t
\\
(\mathit{iv}) \enspace v_{fx}=30\cdot \cos (35)
\\[0.5cm]
\text{3. Resolem els sistema d’equacions:}
\\[0.5cm]
\text{de (iii): }t=\frac{20}{30\cdot \cos (35)}=0.814s\\
\text{i, substituint a (i): }y=40+30\sin (35)\cdot 0.814-4.9{(0.814)}^{2}=\mathrm{50.76}\text{m}
\\[0.5cm]
\text{Quan arribi al terra, hauran transcorregut:}
\\[0.5cm]
0=40+30\sin (35)t-4.9{t}^{2} \rightarrow t=6.27\text{s}
}
\)
3.2 Moviment circular
La trajectòria d’una partícula està formada de moviments lineals i circulars. Segons la llei d’inèrcia de la dinàmica de Newton, per tal de canviar-la, hi ha d’haver una força externa neta que hi actuï. Aquesta força modificarà la direcció o el sentit de la velocitat, però, si és perpendicular, farà que segueixi un camí circular.
Els vectors de velocitat i acceleració centrípeta sempre són tangent i perpendicular a la trajectòria respectivament (l’acceleració centrípeta sempre es dirigeix cap al centre de gir). El mòdul dels vectors és constant, però la direcció d’ambdós canvia constantment.
Quan la distància entre \(P\) i \(P’\) és molt petita, el vector velocitat apunta cap al centre de \(O\) Per tant, com que els triangles isòsceles de velocitats
\(
\displaystyle{
(v_t,v_t,\Delta v)
}
\)
i
\((O, P, P’)\)
són semblants i, considerant que \(s\approx v \cdot \mathrm{\Delta }t\)
\(\displaystyle{
\frac{\mathrm{\Delta }v}{{v}_{t}}\approx \frac{v\cdot \mathrm{\Delta }t}{r}\rightarrow {a}_{c}=\frac{\mathrm{\Delta }v}{t}\approx \frac{{v}_{t}^{2}}{r}}\)
I, quan \(\mathrm{\Delta }t\rightarrow 0,\)aquesta expressió és exacta:
\(\displaystyle{{a}_{c}=\underset{\mathrm{\Delta }t\rightarrow 0}{\lim }\frac{\mathrm{\Delta }v}{\mathrm{\Delta }t}.}\)
Exemple:
\(
\displaystyle{
\text{Dades}
\\[0.5cm]
R=5\mathit{cm},\enspace {v}_{0}=0\text{m/s}, \enspace \alpha =10\text{rad/s},\enspace t=30\text{s}
\\[0.5cm]
\text{1. La velocitat angular, lineal, l’acceleració centrípeta i tangencial despreś de 30 s és:}
\\[0.5cm]
w={w}_{0}+\alpha t=0+10\cdot 30=300\text{rad/s}\rightarrow v=w \cdot R=300 \cdot \mathrm{0,05}=15\text{m/s}
\\
{a}_{c}=\frac{{v}^{2}}{R}=\frac{{15}^{2}}{0.05}=4500m/s\mathrm{{^2}},\alpha ={a}_{t}\cdot R\rightarrow {a}_{t}=\frac{10}{0.05}=200m/s\mathrm{{^2}}
\\[0.5cm]
\text{3. L’angle recorregut després de 30 s és:}
\\[0.5cm]
\theta ={\theta }_{0}+{w}_{0}t+\frac{1}{2}\alpha {t}^{2}=0+0+\frac{1}{2}\cdot 10 \cdot {30}^{2}=4500\text{rad, és a dir,}
\\
4 500\text{rad}\cdot \frac{1\text{volta}}{2\pi \text{rad}}=716\text{ voltes senceres}
}
\)
3.3 Moviment relatiu
3.3.1 Transformacions de Galileu:
Si en un sistema de referència \(S’\) que es mou a velocitat constant respecte a un altre de fix (per exemple, la Terra), una partícula (per exemple un avió) es desplaça del punt \(A\) fins al punt \(B\)
\(
\displaystyle{
\vec{r}=\vec{r}\text{‘}+\vec{u}\cdot t
\\
\frac{d\vec{r}}{\mathit{dt}}=\frac{d\vec{r}\text{‘}}{\mathit{dt}}+\frac{d\vec{u}\cdot t}{\mathit{dt}}
\\
v=v\text{‘}+u
}
\)
\(\displaystyle{\frac{d\vec{r}}{\mathit{dt}}}\) és la velocitat instantània de la partícula mesurada en el sistema de referència \(S\) i \(\displaystyle{\frac{d\vec{r}\text{‘}}{\mathit{dt}}}\)la mesurada en el \(S’\) Per tant, la velocitat d’una partícula amb relació al sistema fix \(S\) és la suma vectorial de la velocitat respecte al \(S’ \)i la relativa de \(S’\) respecte a \(S\)
En mecànica clàssica, el canvi de la velocitat d’una partícula vist des de sistemes de referència diferents és el mateix per a tots els observadors i, en conseqüència, també mesuraran la mateixa acceleració (a=a’):
\(
\displaystyle{
\frac{d\vec{v}}{\mathit{dt}}=\frac{d\vec{v}}{\mathit{dt}}+\frac{d\vec{u}}{\mathit{dt}}
\\
\text{però, com que }\frac{d\vec{u}}{\mathit{dt}}=0\rightarrow \frac{d\vec{v}}{\mathit{dt}}=\frac{d\vec{v}}{\mathit{dt}}\left(a=a\text{‘}\right)
}
\)
Tens dubtes? Vols saber-ne més? T’agradaria que publiquéssim algun tema del teu interès? Has trobat algun error?
Envia’ns un comentari sense compromís i et respondrem tan aviat com ens sigui possible.